PremlataPandit1 and Sankar P Sanyal2
1Indian Institute of Science Education and Research, Bhopal 2Barkatullah University, India
Posters-Accepted Abstracts: J Material Sci Eng
First principle density functional method based on pseufo potential theory is used to investigate the structural, electronic, elastic and phonon properties of Mg2X (X= Si, Ge and Sn) compounds at ambient pressure in their antiflurite structure. The calculated ground state properties such as lattice constants, bulk modulus and its pressure derivatives and the second order elastic constants agree will with the available experimental and other theoretical results. All the above mentioned compounds show, from their band structure calculation, very small indirect band gap. The linear response theory has been applied to calculate the phonon dispersion and density of states. Phonon frequencies are compared with the available Raman modes on these compounds.
Email: sps.physicsbu@gmail.com
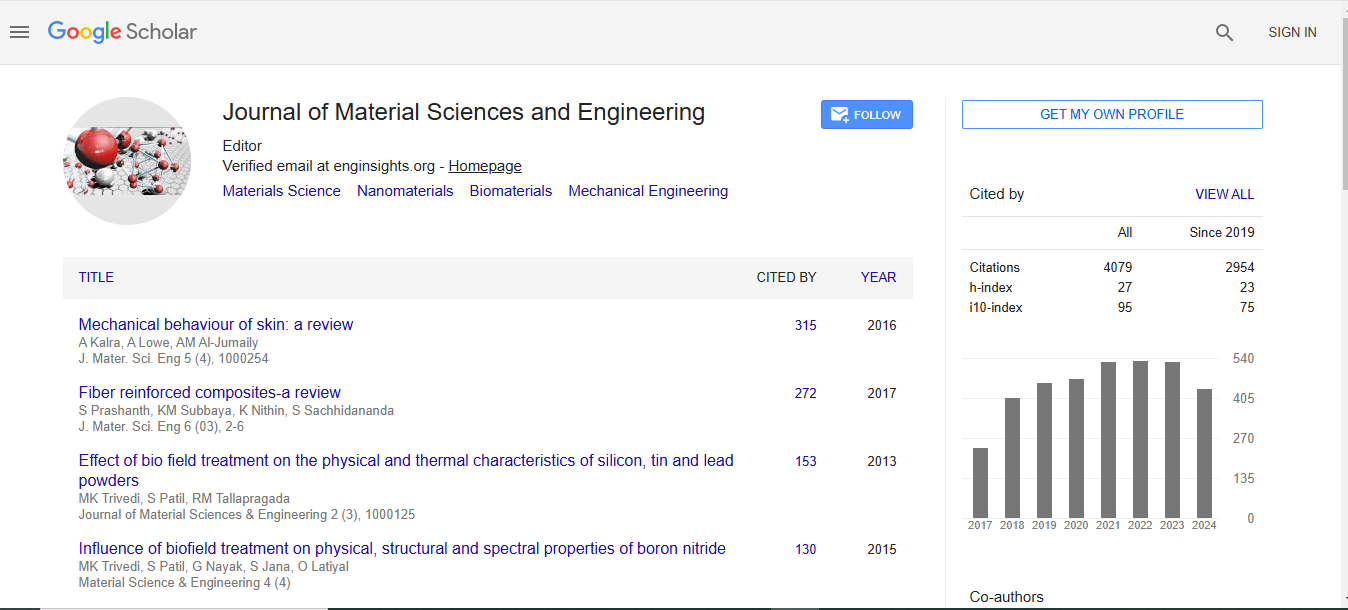
Journal of Material Sciences & Engineering received 3677 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 