Padula MC1, Salvia I1, Mangieri MA1, Busilacchi E2, Martelli G1, Saraceni F2 and Olivieri A2
1Department of Science, University of Basilicata, Potenza, Viale dell��?Ateneo Lucano, 10, 85100, Italy
2Clinic of Hematology, Hospital-University Company ��?Ospedali Riuniti di Ancona�, Ancona, Via Conca, 60126, Italy
Posters-Accepted Abstracts: J Cancer Sci Ther
Introduction. The Wilms��? tumor 1 gene (WT1) is related to several hematopoietic malignancies, including acute myeloid leukemia (AML) [1]. It encodes a 49-52 kDa protein with both oncosuppressor and oncogene roles [2]. WT1 mutational state as AML molecular marker is an attractive research topic for better clarify the gene role in cancer progression [3, 4]. The aim of our study was to investigate the genetic contribution to the AML clinical phenotype searching for WT1 SNPs with functional impact on gene function. Patient and methods. A 80 year-old male was recruited from the Clinic of Hematology, Hospital-University Company ��?Ospedali Riuniti� in Ancona (Italy). Genomic DNA was extracted from patient whole blood by using standard procedures and spectrophotometrically quantified. The whole WT1 gene structure (coding region, exon-intron boundaries, 5��? and 3��?Untranslated Regions) has been screened by means of PCR amplification (with specific primers) and direct sequencing. Bioinformatics software (BlastN and Mutation Surveyor) were used for DNA variant analysis compared to the gene RefSeq (NG_009272.1). In silico investigations were also performed for predicting: RNA fold (RNAfold server), variant deleteriousness (Polyphen2 server), 2D protein structure (Hydrofobic Cluster Analysis, Mobyle server) and 3D protein structure (Phyre2 server). Results. By investigating the mutational state of WT1 gene, as well as minor or known mutations, we found a novel variant, a substitution G>A (NG_009272.1:g.5404G>A, HGSV nomenclature) in homozygosity state. It is responsible, at protein level, for the change p.G70S (NP_000369.3:p.Gly70Ser; HGSV nomenclature) that resides within the WT1 repression domain. Polyphen2 server showed the deleteriousness of the de novo variant. The mRNA folding process occurs in a non-canonical way. The hydrophobic cluster and 3D protein structure were significantly affected by the presence of the variation, that is responsible for the loss of the alpha-helical structure. Conclusion. The molecular findings suggest a functional impairment of WT1 protein that could affect its biological regulatory role. The novel variation could be related to the poor prognosis and provides a significant background for a molecularly-based risk assessment and a subsequent treatment stratification. REFERENCES 1. Owen C, Fitzgibbon J and Paschka P. 2010. The clinical relevance of Wilms Tumour1 (WT1) gene mutations in acute leukaemia. Hematol Oncol. 28: 13-19 2. Yang L, Han Y, Suarez Saiz F and Minden MD. 2007. A tumor suppressor and oncogene: The WT1 story. Leukemia. 21: 868- 876. 3. Borate U, Absher D, Erba HP and Pasche B. 2012. Potential of whole-genome sequencing for determining risk and personalizing therapy: focus on AML. Expert Rev Anticancer Ther. 12(10): 1289-1297. 4. Link DC. 2012. Molecular genetics of AML. Best Pract Res Clin Haematol. 25(4): 409-414.
Email:
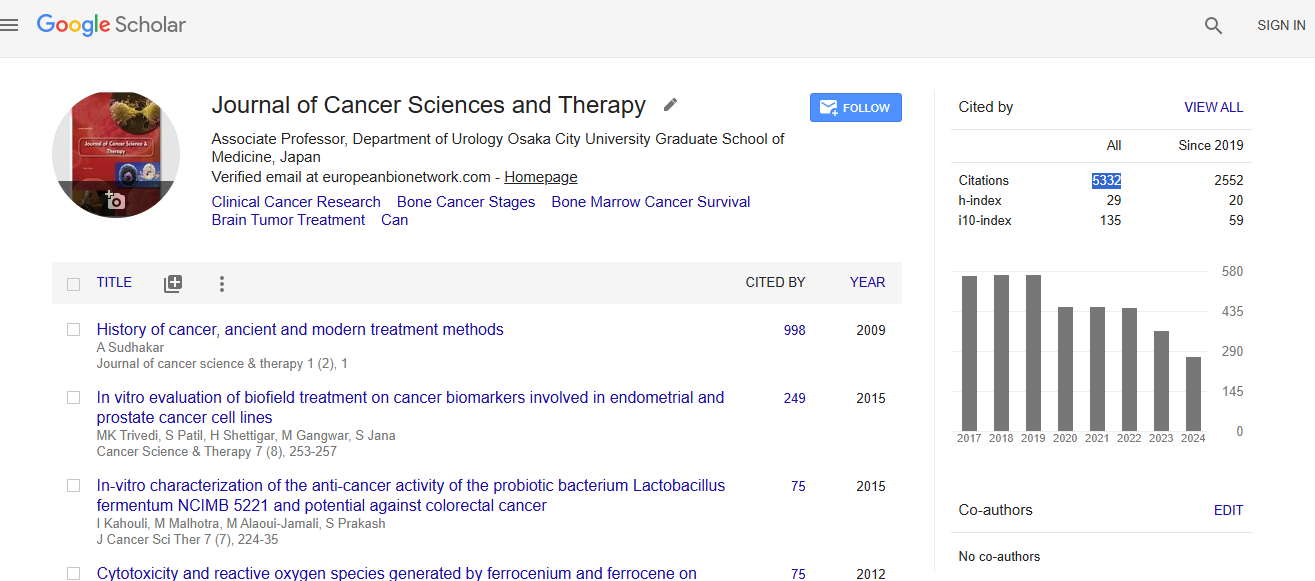
mcpadula25@gmail.comCancer Science & Therapy received 5332 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 