Muhammad Mahajnah and Rajech Sharkia
Hillel-Yaffe Medical Center, Israel
The Triangle Regional Research and Development Center, Isra
Scientific Tracks Abstracts: J Neurol Disord
About half of patients with rare movement disorders such as hereditary spastic paraplegias and cerebellar ataxias remain
genetically unexplained, implicating novel genes and unrecognized mutations in known genes. Non-coding DNA variants
are suspected to account for a substantial part of undiscovered causes of rare diseases. Whole-exome sequencing findings
in a recessive spastic ataxia family turned our attention to intronic variants in POLR3A, a gene previously associated with
hypomyelinating leukodystrophy type 7. We screened a cohort of hereditary spastic paraplegia and cerebellar ataxia cases
(n=618) for mutations in POLR3A and identified compound heterozygous POLR3A mutations in â�?¼3.1% of index cases.
Interestingly, >80% of POLR3A mutation carriers presented the same deep-intronic mutation (c.1909+22G>A), which
activates a cryptic splice site in a tissue and stage of development-specific manner and leads to a novel distinct phenotype. The
phenotype is characterized by adolescent-onset progressive spastic ataxia with frequent occurrence of tremor, involvement of
the central sensory tracts and dental problems (hypodontia, early onset of severe and aggressive periodontal disease). Instead of
the typical hypomyelination magnetic resonance imaging pattern associated with classical POLR3A mutations, cases carrying
c.1909+22G>A demonstrated hyperintensities along the superior cerebellar peduncles. These hyperintensities may represent the
structural correlate to the cerebellar symptoms observed in these patients. We demonstrate that autosomal-recessive mutations
in POLR3A are a frequent cause of hereditary spastic ataxias, accounting for about 3% of hitherto genetically unclassified
autosomal recessive and sporadic cases; hypomyelination is frequently absent in POLR3A-related syndromes, especially when
intronic mutations are present, and thus can no longer be considered as the unifying feature of POLR3A disease. Our results
demonstrate that substantial progress in revealing the causes of Mendelian diseases can be made by exploring the non-coding
sequences of the human genome..
Recent Publications
1. Azmanov D N, Siira S J, Chamova T, Kaprelyan A, Guergueltcheva V, Shearwood AJ, et al (2016) Transcriptome-wide
effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum Mol Genet.
25:4302��?14.
2. Schule R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S, et al (2016) Hereditary spastic paraplegia -clinico-genetic
lessons from 608 patients. Ann Neurol 79:646��?58.
3. Cayami F K, La Piana R, Van Spaendonk R M, Nickel M, Bley A,Guerrero K, et al (2015) POLR3A and POLR3B
mutations in unclassified hypomyelination. Neuropediatrics 46:221��?8.
4. Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, et al (2015) Exome sequencing in undiagnosed
inherited and sporadic ataxias. Brain 138(2):276��?83.
5. Terao Y, Saitsu H, Segawa M, Kondo Y, Sakamoto K, Matsumoto N, et al (2012) Diffuse central hypomyelination
presenting as 4H syndrome caused by compound heterozygous mutations in POLR3A encoding the catalytic subunit
of polymerase III. J Neurol Sci 320:102��?5.
Muhammad Mahajnah is an Assistant Professor of Pediatrics and Pediatric Neurology at the Technion Faculty of Medicine, Israel. He completed his Medical degree at Bruce and Ruth Rappaport Faculty of Medicine, Technion, Israel in 1992 and his PhD degree in 1998. He trained in Pediatrics at Carmel Medical Center and completed fellowship in Pediatric Neurology at Schneider Children Medical Center, Tel Aviv. He has worked with children neurological disorder for about 20 years and has special interest in neurodevelopmental disorders and neuro genetic disorders and devotes his time to both clinical work and research.
E-mail: mohamedm@hy.health.gov.il
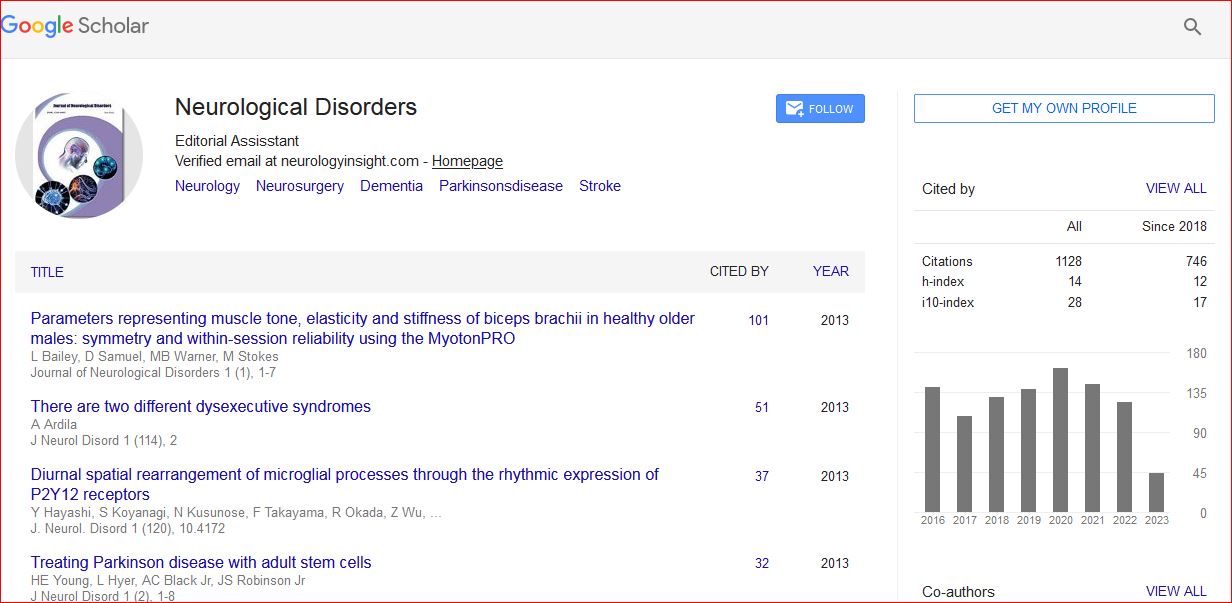
Neurological Disorders received 1343 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 