Mazharul M Islam
Universit�?¤t Bonn, Germany
Scientific Tracks Abstracts: J Material Sci Eng
Li-ion batteries (LIBs) have a great combination of high energy and power density capacities, which has made the LIB technology as the prime choice for portable electronics, power tools, and hybrid/full electric vehicles. The electrochemical performance of LIBs strongly depends on the electrode properties. The conventional Li-ion batteries use Li metal and carbonbased materials, typically graphite, as anode materials. However their applications are limited due to safety issues caused by dendrites formation. Therefore alternative anode materials are employed for the next generation LIBs which cover titanium oxide based materials as well as alloying/de-alloying materials with Si. In the present contribution, the stoichiometric and defect properties of lithium titanim oxides and various LixSiy compounds are investigated theoretically using the first-principles density functional theory (DFT) methods. We have considered two different Li2Ti3O7 structures: ramsdellite and layered types. In ramsdellite Li2Ti3O7 Li+ can migrate along the ��?one dimensional channel��? or along crystallographic ac plane. Our calculated EA for Li+ diffusion in the one dimensional channel ranges from 0.20��?0.92 eV and that in the ac plane ranges from 0.30��?0.85 eV. However due to the large energy difference between the initial and final stages for Li diffusion in the case of ac plane shows that these pathways are kinetically prohibited. Therefore, the ramsdellite Li2Ti3O7 is a qasi-one-dimensional ion conductor. In the layered Li2Ti3O7, Li migrates along the crystallographic b direction by a vacant tetrahedral cite as well as along the ab plane in a direct pathway. The ab-plane migration is the most likely as it has a smaller activation barrier (0.4 eV) compared to the tetrahedral migration pathway (0.95 eV). Recent studies have provided evidence for the formation of various stable Li silicide crystalline phases such as LiSi, Li4.7Si2, Li12Si7, Li7Si3, Li13Si4, Li15Si4, and Li22Si5 during lithiation. In the present study, we have studied LiSi and Li13Si4 in order to search for a fast Li ion conductor. Our study shows that the interlayer interactions due to the van der Waals (vdW) forces play a key role for the structure and energetic properties of LixSiy materials. In case of LiSi, Li migrates to the 1st and 2nd nearest distance by a Li point defect. The activation energy for the both possible pathways range between 0.2 to 0.25 eV, indicating a very fast ion conduction in LiSi compounds. On the other hand, Li migration in Li13Si4 is slightly more complex. Li migrates either in a direct migration pathway or by a tetrahedral pathway. The activation energy for the direct migration pathway is 0.65 eV whereas that for the tetrahedral pathway is 0.75 eV. Therefore we conclude that Li13Si4 is slower ionic conductor than LiSi.
Dr. Mazharul M Islam has been working as senior research fellow at the Mulliken Center for Theoretical Chemistry of the University of Bonn Germany. He has got the expertise on theoretical modeling using various first principles DFT approaches. His interest of research activities has covered a wide variety of areas such as the ion conductivity in solid state materials having applications in green energy sources, the investigation of photocatalytic activities of TiO2 due to its various industrial applications, the modeling of basic mechanisms of corrosion processes of metallic materials during their applications in construction, power generation etc, and the characterization of mesoporous oxide supported metal oxide catalysts having applications in industries. He was awarded with couple of honors and awards, various funding and huge number of collaborations in his career. He has published 3 scholarly books, 2 scholarly book chapters and 53 articles in peer reviewed journals so far.
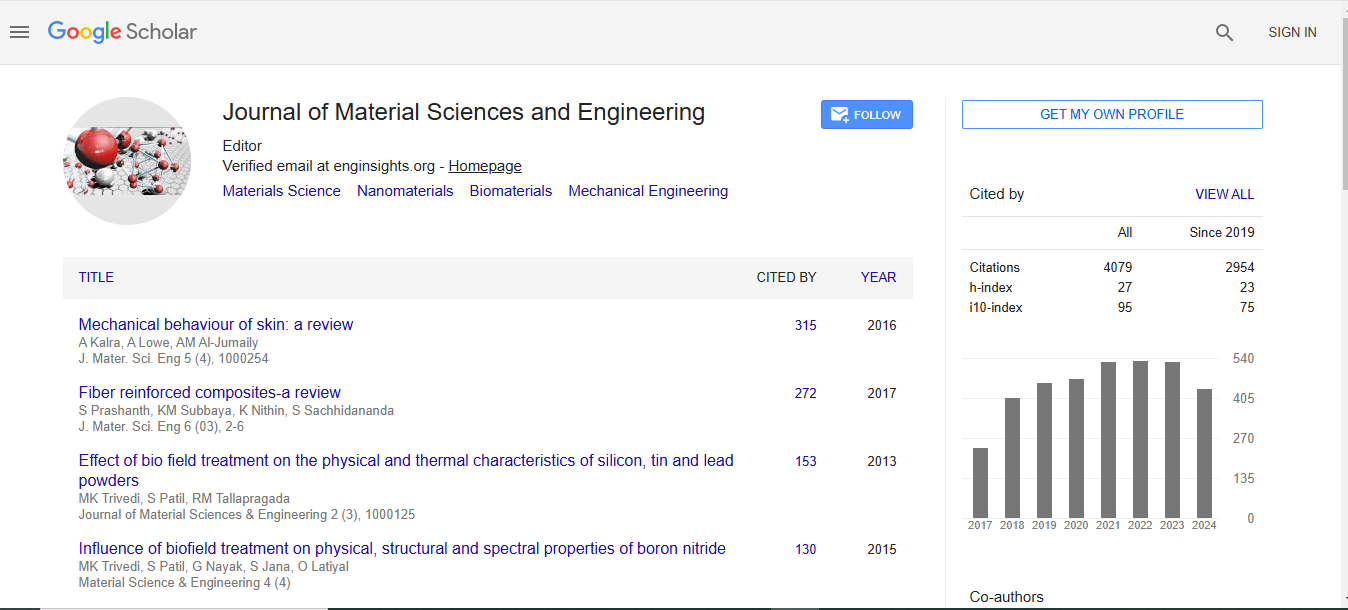
Journal of Material Sciences & Engineering received 3677 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 