Michael Leffak
Wright State University, USA
Scientific Tracks Abstracts: Mol Biol
Short repetitive DNA sequences, termed microsatellite DNAs, comprise about 3% of human DNA and are widely distributed across the genome. The self-interacting nature of many microsatellites makes them susceptible to non-Watson-Crick structure formation. These repetitive sequences and their noncanonical structures impede DNA replication and repair and make microsatellite replication error-prone. Indeed, expansion of microsatellite repeat tracts during replication or repair is responsible for more than thirty neurodegenerative diseases. In contrast to microsatellite expansions, the presence of tandem repeats at chromosomal breakpoints is also consistent with the hypothesis that replication stalling at these structures exposes DNA to nuclease attack and replication-dependent DNA double-strand breaks (DSBs). To test this hypothesis, we developed a dual fluorescence (DF) reporter assay to analyze DSBs in vivo at (CTG/CAG) trinucleotide repeat and (Pu/Py) mirror repeat microsatellites. In this system, microsatellites are integrated at a single chromosomal site in HeLa cells, bordered by the c-myc replication origin core, an I-SCE1 site, and two fluorescent protein marker genes flanked by AluY elements. I-SCE1 cleavage alongside the microsatellites leads to homologous recombination between AluY elements and loss of one of the marker genes, which can be detected by flow cytometry. However, in the absence of I-SCE1 cleavage, we find that these microsatellites are also sensitive to DSBs from endogenous factors. Strikingly, �??clean�?� DSBs generated by I-SCE1 digestion in vivo are repaired by homologous recombination (HR), but microsatellite replication-dependent DSBs are refractory to canonical HR. In addition, exogenous replication stressors significantly enhance the frequency of DSBs at the (CTG/CAG)100 and asymmetric (Pu/Py) repeats. Thus, replication-dependent DSBs at these repeats are contingent on microsatellite length and replication polarity, respectively. Knockdowns of DNA damage response proteins increase or decrease the sensitivity of the (CTG/CAG) 100 microsatellites to replication stress, and implicate postreplication repair in the generation of DSBs. The unique structure of these single-ended DSBs leads to error-prone break-induced replication (BIR). BIR has been implicated in the clustered point mutations and gross chromosomal rearrangements (GCRs) observed in human tumors. Our results show that, as in yeast models, BIR from the (CTG/CAG)100 microsatellites is highly mutagenic and leads to GCRs.
Dr Michael Leffak is a Professor of Biochemistry and Molecular Biology at the Boonshoft School of Medicine, Wright State University, Dayton, Ohio, USA. His laboratory discovered the c-myc origin of DNA replication and continues to use this functionality to identify proteins involved in replication initiation; to characterize the consequences of DNA replication on microsatellite DNA sequences, and to identify the effects of non-Watson-Crick microsatellite DNA structures on DNA breaks and replication-dependent genomic instability.
E-mail: michael.leffak@wright.edu
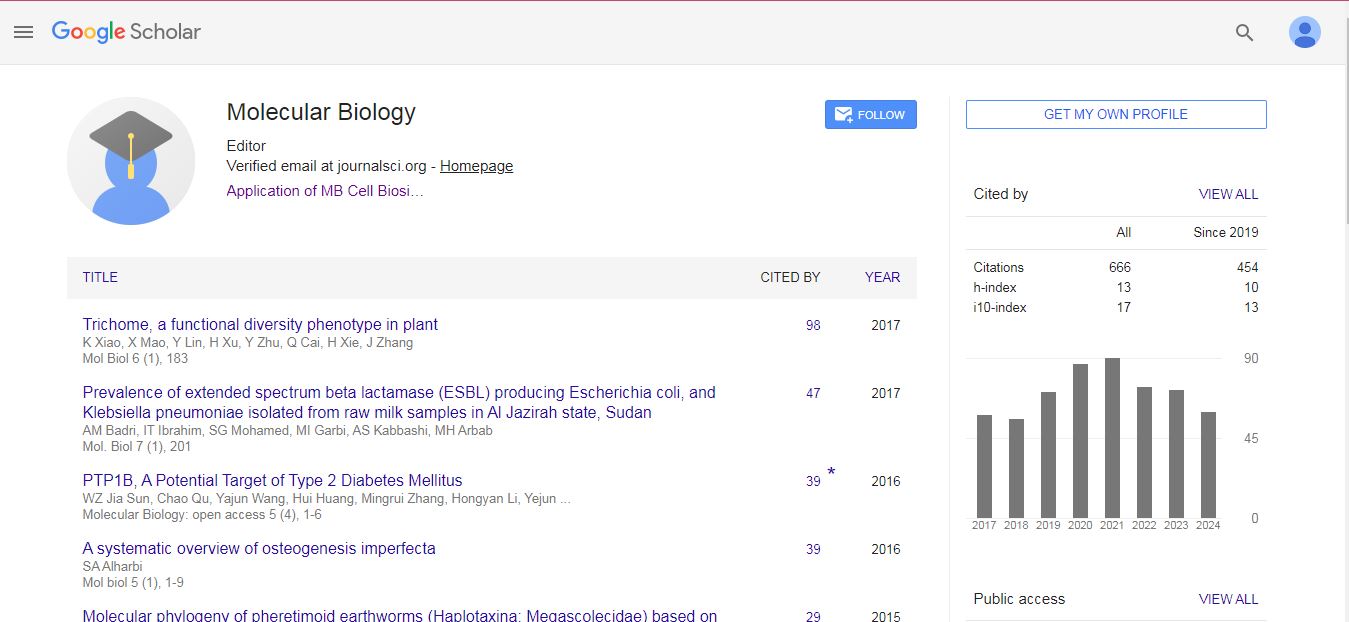
Molecular Biology: Open Access received 607 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 