Editorial
Pages: 0 - 0Erin L Abner, Richard J Charnigo and Richard J Kryscio
DOI:
DOI: 10.4172/2155-6180.S1-e001
A variety of statistical methods are available to investigators for analysis of time-to-event data, often referred to as survival analysis. Kaplan-Meier estimation and Cox proportional hazards regression are commonly employed tools but are not appropriate for all studies, particularly in the presence of competing risks and when multiple or recurrent outcomes are of interest. Markov chain models can accommodate censored data, competing risks (informative censoring), multiple outcomes, recurrent outcomes, frailty, and non-constant survival probabilities. Markov chain models, though often overlooked by investigators in time-to-event analysis, have long been used in clinical studies and have widespread application in other fields.
Research Article
Pages: 1 - 9Xiao-Lin Wu, Daniel Gianola, Zhi-Liang Hu and James M. Reecy
DOI:
Meta-analysis is an important method for integration of information from multiple studies. In quantitative trait association and mapping experiments, combining results from several studies allows greater statistical power for detection of causal loci and more precise estimation of their effects, and thus can yield stronger conclusions than individual studies. Various meta-analysis methods have been proposed for synthesizing information from multiple candidate gene studies and QTL mapping experiments, but there are several questions and challenges associated with these methods. For example, meta-analytic fixed-effect models assume homogeneity of outcomes from individual studies, which may not always be true. Whereas random-effect models takes into account the heterogeneity among studies they typically assume a normal distribution of study-specific outcomes. However in reality, the observed distribution pattern tends to be multi-modal, suggesting a mixture whose underlying components are not directly observable. In this paper, we examine several existing parametric meta-analysis methods, and propose the use of a non-parametric model with a Dirichlet process prior (DPP), which relaxes the normality assumptions about study- specific outcomes. With a DPP model, the posterior distribution of outcomes is discrete, reflecting a clustering property that may have biological implications. Features of these methods were illustrated and compared using both simulation data and real QTL data extracted from the Animal QTLdb (http://www.animalgenome.org/cgi-bin/QTLdb/index). The meta analysis of reported average daily body weight gain (ADG) QTL suggested that there could be from six to eight distinct ADG QTL on swine chromosome 1
Research Article
Pages: 1 - 9DOI:
The Bayesian model selection approach has been adopted by more and more people when analyzing a large data. However, it is known that the reversible jump MCMC (RJMCMC) algorithm, which is perhaps the most popular MCMC algorithm for Bayesian model selection, is prone to get trapped into local modes when the model space is complex. The stochastic approximation Monte Carlo (SAMC) algorithm essentially overcomes the local trap problem suffered by conventional MCMC algorithms by introducing a self-adjusting mechanism based on the past samples. In this paper, we propose a population SAMC (Pop-SAMC) algorithm, which works on a population of SAMC chains and can make use of crossover operators from genetic algorithms to further improve its efficiency. Under mild conditions, we show the convergence of this algorithm. Comparing to the single chain SAMC algorithm, Pop-SAMC provides a more efficient self-adjusting mechanism and thus can converge faster. The effectiveness of Pop-SAMC for Bayesian model selection problems is examined through a change-point identification problem and a gene selection problem. The numerical results indicate that Pop-SAMC significantly outperforms both the single chain SAMC and RJMCMC.
Research Article
Pages: 1 - 7Kris M Jamsen, Sophie G Zaloumis, Katrina J Scurrah and Lyle C Gurrin
DOI:
Statistical models imposed on family data can be used to partition phenotypic variation into components due to sharing of both genetic and environmental risk factors for disease. Generalized linear mixed models (GLMMs) are useful tools for the analysis of family data, but it is not always clear how to specify individual-level regression equations so that the resulting within-family variance-covariance matrix of the phenotype reflects the correlation implied by the relatedness of individuals within families. This is particularly challenging when families are of varying sizes and compositions. In this paper we propose a general approach to specifying GLMMs for family data that uses a decomposition of the within-family variance-covariance matrix of the phenotype to set up a series of regression equations with fixed and random effects that corresponds to an appropriate genetic model. This “mechanistic” specification is particularly suited to estimation and evaluation of models within a Markov chain Monte Carlo (MCMC) framework. The proposed approach was assessed with simulated data to demonstrate the accuracy of estimation of the variance components. We analyzed data from the Victorian Family Heart Study (families with two generations over-sampled for those with monozygotic and dizygotic twins) and for a binary phenotype (hypertension) that resulted in substantially reduced computation time in the MCMC framework (via WinBUGS) compared with a maximum likelihood approach (via Stata). The proposed approach to model specification in this paper is easily implemented using standard software and can accommodate prior information on the magnitude of fixed or random effects.
Research Article
Pages: 1 - 6DOI:
Abstract
Background and objective: New analytical tools are needed to advance tobacco research, tobacco control planning and tobacco use prevention practice. In this study, we validated a method to extract information from crosssectional survey for quantifying population dynamics of adolescent smoking behavior progression.
Methods: With a 3-stage 7-path model, probabilities of smoking behavior progression were estimated employing the Probabilistic Discrete Event System (PDES) method and the cross-sectional data from 1997-2006 National Survey on Drug Use and Health (NSDUH). Validity of the PDES method was assessed using data from the National Longitudinal Survey of Youth 1997 and trends in smoking transition covering the period during which funding for tobacco control was cut substantively in 2003 in the United States.
Results: Probabilities for all seven smoking progression paths were successfully estimated with the PDES method and the NSDUH data. The absolute difference in the estimated probabilities between the two approaches varied from 0.002 to 0.076 (p>0.05 for all) and were highly correlated with each other (R2=0.998, p<0.01). Changes in the estimated transitional probabilities across the 1997-2006 reflected the 2003 funding cut for tobacco control.
Conclusions: The PDES method has validity in quantifying population dynamics of smoking behavior progression with cross-sectional survey data. The estimated transitional probabilities add new evidence supporting more advanced tobacco research, tobacco control planning and tobacco use prevention practice. This method can be easily extended to study other health risk behaviors.
Review Article
Pages: 1 - 13DOI:
In this article, we present a selective overview of some recent developments in Bayesian model and variable selection methods for high dimensional linear models. While most of the reviews in literature are based on conventional methods, we focus on recently developed methods, which have proven to be successful in dealing with high dimensional variable selection. First, we give a brief overview of the traditional model selection methods (viz. Mallow’s Cp, AIC, BIC, DIC), followed by a discussion on some recently developed methods (viz. EBIC, regularization), which have occupied the minds of many statisticians. Then, we review high dimensional Bayesian methods with a particular emphasis on Bayesian regularization methods, which have been used extensively in recent years. We conclude by briefly addressing the asymptotic behaviors of Bayesian variable selection methods for high dimensional linear models under different regularity conditions.
Research Article
Pages: 1 - 7DOI:
Accurate estimates of disease risk (penetrance) associated with inherited gene mutations are critical for the clinical management of individuals at risk, but this estimation raises many statistical challenges especially when performed in a family-based design. In this paper, we propose a general frailty model-based approach to accommodate this design, where the frailty random effect accounts for shared risk among family members not due to the observed risk factors. It is of major interest when the goal is to discover other genetic variations besides the major gene and to get accurate estimates of penetrance (i.e. unbiased by unknown confounding factors). This approach is further extended to accommodate missing genotypes in family members and the non-random ascertainment of the families. Simulation results show that the proposed method performs well in realistic settings. Finally, a family-based breast cancer study of the BRCA1 and BRCA2 genes is used to illustrate the method.
Research Article
Pages: 1 - 4Changchun Xie, Xuewen Lu and Janice Pogue
DOI:
The Attributable Fraction or risk function (ARF) is used to measure the impact of an exposure on occurrence of disease within a population. For any prospective cohort study, risk is likely to be estimated using time to event or survival data. Attributable risk function with right censored survival data has been discussed by Samuelsen and Eide. We propose a natural extension of the ARF to clustered survival data, which are common in medical research. We derive an estimator of the ARF. Simulation studies are conducted to evaluate the performance of our method and investigate the consequences of ignoring the cluster effect in analysis.
Research Article
Pages: 1 - 6DOI:
In the analysis of recurrent event data, frailties are commonly used to model the dependence structure among repeated event times within an individual. Often it is of interest to test whether the variance component in a frailty model is zero. It is well-known that the usual asymptotic mixtures of chi-square distributions of the score statistics for testing constrained variance components do not necessarily hold. In this paper, we propose and explore a stochastic permutation score test based on randomly permuting the indices associated with the individuals of a survival model. An empirical study suggests that the proposed score test has approximately the correct level of significance and is more powerful than the asymptotic score test based on the mixture of chi-square distributions. The proposed test is illustrated using two sets of actual recurrence failure time data obtained from clinical experiments.
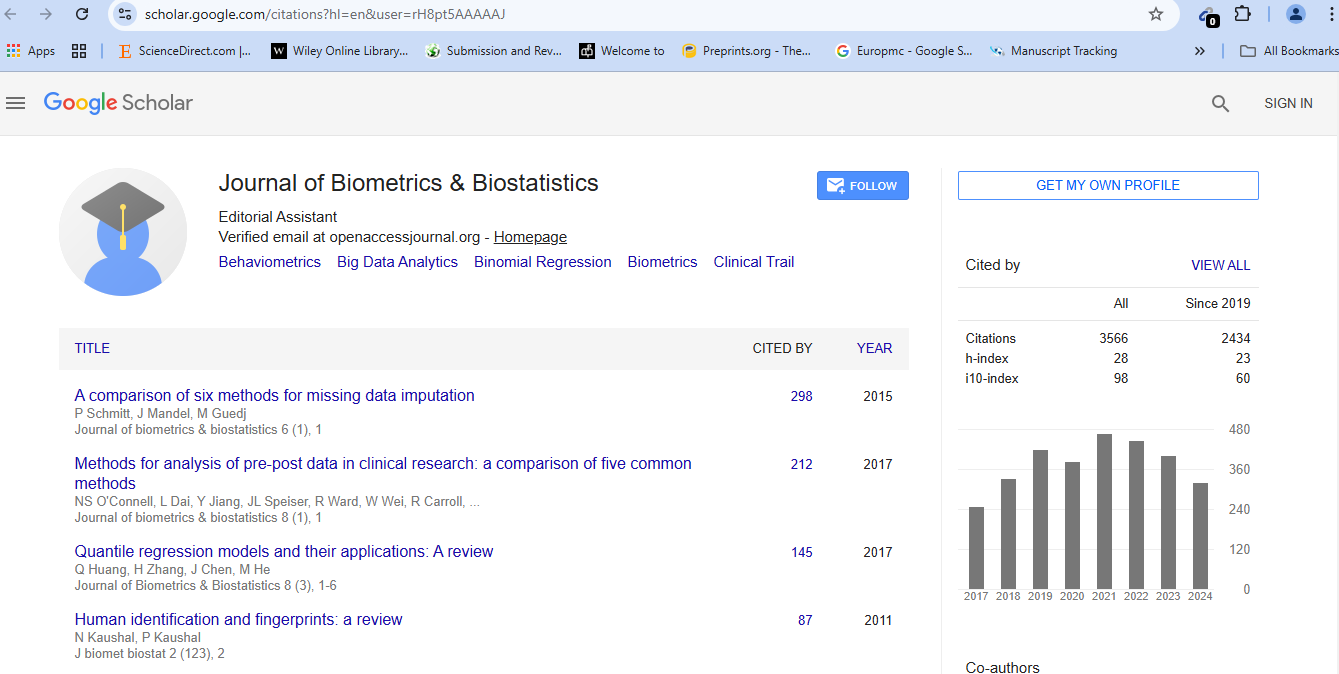
Journal of Biometrics & Biostatistics received 3496 citations as per Google Scholar report
 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 