Editorial
Pages: 0 - 0Derek S Young, Limin Feng and Richard J Charnigo
Research Article
Pages: 0 - 0DOI:
DOI: 10.4172/2155-6180.S12-001
The use of joint models that are capable of handling different data types is becoming increasingly popular in biomedical practice. Evaluation of various statistical techniques that have been developed for mixed data in simulated environments requires concurrent generation of multiple variables. In this article, I comprehensively evaluate the unified framework proposed by Demirtas et al. for simultaneously generating binary and nonnormal continuous data given the marginal characteristics and correlation structure. I conduct this assessment in three simulated settings with synthetic bivariate and multivariate data as well as real depression score data from psychiatric research. Considering close agreement between the specified and empirically computed quantities on average, as measured by some key bias- and precision-related quantities, the methodology appears to have prospects to address the need of generating intensive data that have binary and continuous parts for simulation purposes.
Research Article
Pages: 0 - 0Yuejiao Fu, Pengfei Li and Soowoon Chung
DOI:
DOI: 10.4172/2155-6180.S12-002
Mixture models provide flexible means of handling heterogeneity in data. The possible latent structure suggested by mixture model analysis should be carefully examined using designed experiments. Sample size determination is an important and difficult step in design of experiments. We investigate the sample size calculation for the modified likelihood ratio test for binomial mixture models arising in genetic linkage analysis. We obtain limiting distributions for the modified likelihood ratio test under two sets of commonly used local alternatives. A simple sample-size formula is obtained and illustrated using both simulations and a genetic linkage study for schizophrenia.
Research Article
Pages: 0 - 0Rhonda D Szczesniak, Kert Viele and Robin L Cooper
DOI:
DOI: 10.4172/2155-6180.S12-003
A shape invariant model for functions f1,…,fn specifies that each individual function fi can be related to a common shape function g through the relation fi(x)=aig(cix + di) + bi. We consider a flexible mixture model that allows multiple shape functions g1,…,gK, where each fi is a shape invariant transformation of one of those gk. We derive an MCMC algorithm for fitting the model using Bayesian Adaptive Regression Splines (BARS), propose a strategy to improve its mixing properties and utilize existing model selection criteria in semiparametric mixtures to select the number of distinct shape functions. We discuss some of the computational difficulties that arise. The method is illustrated using synaptic transmission data, where the groups of functions may indicate different active zones in a synapse.
Research Article
Pages: 0 - 0DOI:
DOI: 10.4172/2155-6180.S12-004
Image registration is one of the fundamental tasks within image processing. It has wide applications in the fields of medical imaging, computer vision, statistical modeling etc. It is required when one wants to combine valuable statistical information from multiple images, possibly acquired using different modalities, at different time points or from different subjects, or to compare or integrate the data obtained from same or different measures. The subject of this article is nonrigid image registration. In particular, the main focus is on the application of fluid dynamics and mutual information (MI), and the comparison of two different similarity measures, i.e. mutual information and sum of squared differences (SSD). Numerical experiments show that fluid registration using SSD is ideal for mono-modal image registration, while fluid registration using MI does a better job in multi-modal image registration.
Research Article
Pages: 0 - 0Qian Fan, Richard Charnigo, Zohreh Talebizadeh and Hongying Dai
DOI:
DOI: 10.4172/2155-6180.S12-005
In this work we consider a three-component normal mixture model in which one component is known to have mean zero and the other two contaminating components have a nonnegative and a no positive mean respectively, while all three components share a common unknown variance parameter. One potential application of this model may be in prioritizing statistical scores obtained in biological experiments, including genetics data. Such a mixture model may be useful in describing the distribution of numerous Z test statistics corresponding to different genes or SNPs, such that a “significant” Z test statistic for a particular gene suggests its connection to a medical condition. More specifically, the inferences drawn from such a mixture model may be useful in a filtration algorithm to remove large subsets of genes or SNPs from consideration, thereby reducing the need for stringent and power-depleting multiplicity adjustments for controlling type I error rates on the remaining genes. We show how to test whether there is contamination in at least one direction (i.e., the mixture model truly requires at least two components) and, if so, how to test whether there is contamination in both directions (i.e., the mixture model truly requires all three components). We assess our testing procedures in simulation studies and illustrate them through application to LOD scores in a genome-wide linkage analysis from an autism study.
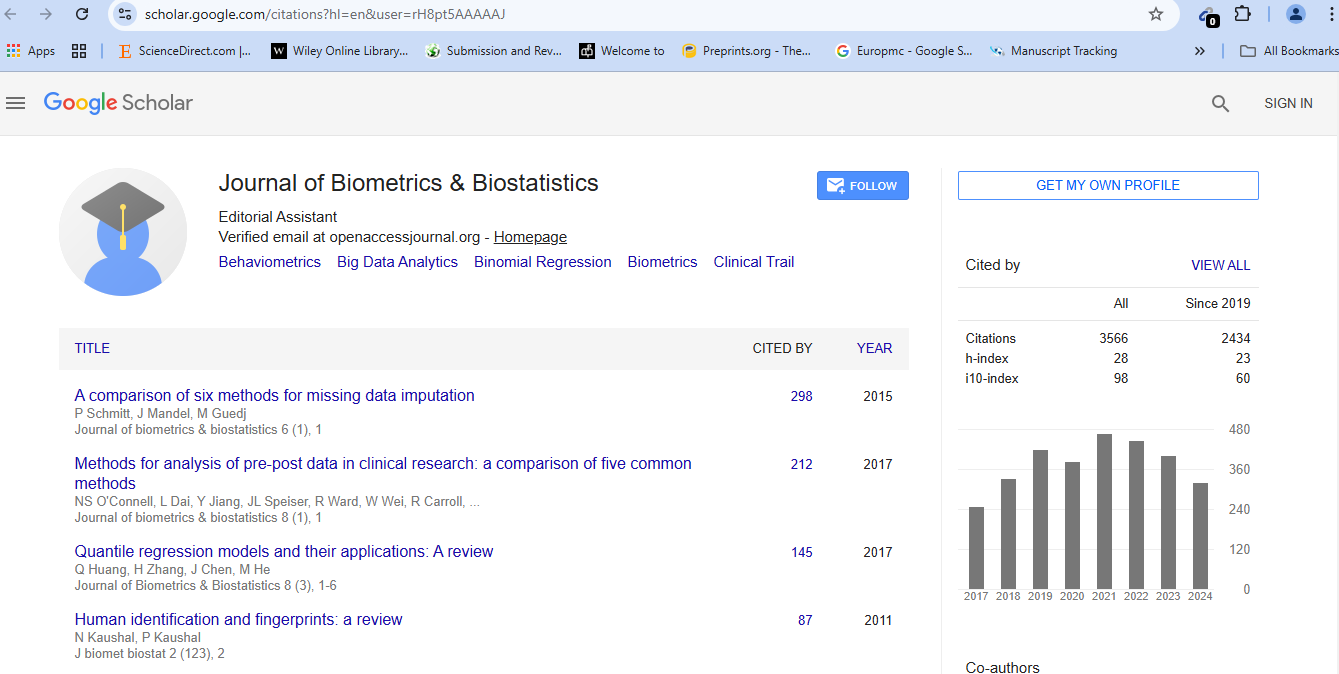
Journal of Biometrics & Biostatistics received 3496 citations as per Google Scholar report
 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 